

Trainee Spotlight: Jittasak (Jake) Khowsathit, PhD
-
Jittasak (Jake) Khowsathit, PhD
Postdoctoral Associate
Dr. John Karanicolas' Lab
Fox Chase Cancer Center
[email protected]Biography
I have always been interested in science. “Dexter’s Laboratory” and “Pinky and the Brain” were amongst my favorite cartoons when I was little. During high school I had an opportunity to join Thailand’s Junior Science Talent Project, where they fund research labs across the country to take in high schoolers to do real research. That’s how I ended up studying Rickettsia spp., well known to cause Rocky Mountain Spotted fever here in the US. During my undergrad years at Chulalongkorn University, I switched from studying pathogens to studying shrimp immunity (shrimp is one of the major food exports in Thailand and is very important for the economy). It was during these years that I developed a fascination for proteins and their role in life. During one of my classes, a professor mentioned in his lecture that “proteins were designed by mother nature and we could only mimic her work, but never create anything novel.” I was determined to prove him wrong; that’s when I knew I wanted to study novel protein design.
My path brought me to John Karanicolas’ lab at the University of Kansas. He was starting to explore the use of computational approaches (Rosetta) to design novel protein switches where the protein’s function could be turned “on and off” using small molecules. In the beginning, we sought to demonstrate that the idea worked as a proof of concept. Then one day John called me into his office with a poker face. I thought I was in trouble. He said, “The lab will be moving to Fox Chase Cancer Center, are you interested in tagging along?” After driving a giant U-Haul truck for 1,200 miles, we ended up here in Philly in 2016. Aligning my previous work with Fox Chase’s goal, we decided to apply our computational approaches to design novel cancer treatments. Based on this work, I completed my Ph.D. in 2019. As a postdoc at FCCC, I am using the methods developed in my graduate work to create immune-checkpoint inhibiting antibodies that have fewer side effects than currently available therapies.
Research Overview
Therapeutic monoclonal antibodies have transformed medicine, particularly for use in cancer treatment. More than 50 antibody-derived drugs have already reached the clinic, the majority of which target cytokines or cell-surface receptors. Unfortunately, many of these targets have adverse side effects caused by “on-target off-tumor” activities. To mitigate this, we have developed a strategy whereby an antibody’s ability to recognize its target is controlled by a small molecule. We envisioned that the designed-antibody will be administered in an “inactive” form and will be activated only in the presence of the rescuing small molecule. The small molecule will be administered locally to the tumor. Thus, antibody activation (and toxicity) only occurs in the tumor microenvironment.
To design pairs of inactivated antibodies and rescuing small molecules, we developed a novel computational strategy. First, we model a set of mutated versions of the antibody. Each mutated version has a uniquely-shaped cavity on the interface of the molecule, causing the antibody to be nonfunctional. Then, we carry out a complementary virtual screen to identify drug-like compounds that bind in each candidate cavity, thus restoring the function of the antibody. Based on our models, we identified the most compatible antibody/rescue molecule pairs to test in vivo. We demonstrated the utility of this strategy in a fluorescein-binding single-chain variable fragment (scFv) antibody and experimentally characterized a triple mutant with reduced antigen-binding (Rip-3) that can be rescued using a complementary small molecule (Stitch-3).
The residues that were mutated to generate the Rip-3 fragment are conserved in many therapeutic antibodies, suggesting that the Rip-3/Stitch-3 (mutation/small molecule) pair may serve as a general starting point for introducing small molecule-dependence into many clinically-relevant antibodies. Currently, we are in the process of applying what we have learned in our model system to immune checkpoint inhibiting antibodies (anti-PD-1, anti-PD-L1, and anti-CTLA-4). Successful implementation of small molecule-dependent antibodies in the clinic may lead to reduced immune-related adverse events associated with current treatments.
Featured Publication
Khowsathit J, Bazzoli A, Cheng H, Karanicolas J. Computational Design of an Allosteric Antibody Switch by Deletion and Rescue of a Complex Structural Constellation. ACS Central Science. 2020 6 (3), 390-403. DOI: 10.1021/acscentsci.9b01065
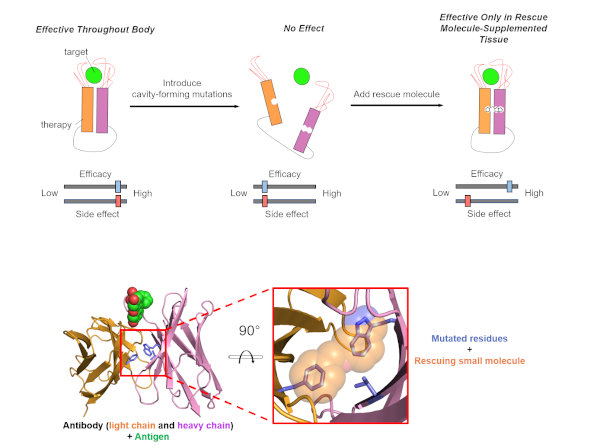
Figure 1. Designing small molecule-dependent antibodies to reduce side effects of antibody therapies. (Top) Summary of the design strategy. Introducing cavity-forming mutations (Rip-3) in the interface between the heavy and light chains of a therapeutic antibody will lead to dissociation of this domain−domain interface, leading to loss of antigen-binding activity. Subsequent addition of a rescuing small molecule (Stitch-3) will induce reassociation of the domain−domain interface and, thus, restore activity. (Bottom) Model of the rescued Rip-3/Stitch-3 complex. The residues that comprise the Rip-3 constellation are indicated in blue sticks and are shown in superposition with the small molecule predicted to rescue this mutation (Stitch-3, orange). Green spheres represent the antigen (fluorescein).